PART 2: Blood Cells & Long Covid
With a foundation in the S1 sui pathogenesis we will now look at the blood, immune cells, and Long Covid. Chronic conditions may be the result of secondary pathologies. Science must always begin with natural observations to gain an understanding of physiological mechanisms related to their existence. With understanding comes methodologies of clinical and/or diagnostic confirmation.
Long-Term Changes to Blood Cells
Scientists have observed significant changes in lymphocyte stiffness, monocyte size, neutrophil size and deformability, and heterogeneity of erythrocyte deformation and size. Although some of these changes recovered to normal values after hospitalization, others persisted for months after hospital discharge, evidencing the long-term imprint of COVID-19 on the body.
Change in physical form may happen at the phospho-lipid bilayer cell membrane like TER, “inflammation”, a mutation in genetics, or a change in microtubule (tubulin) genetic expression and natural function. The authors hypothesize a “cytoskeleton, responsible for determining cell function, has changed”; likely referencing microtubule (tubulin) structures.

COVID-19 Causes Long-Term Blood Cell Changes
https://www.sciencedirect.com/science/article/pii/S0006349521004549?via%3Dihub
‘We suspect that the immune cells’ cytoskeleton, which is responsible for determining cell function, has changed,’ explains Markéta Kubánková, lead author of the research article. In her opinion, real-time deformability cytometry could potentially be used routinely for diagnosing Covid-19, and even act as an early warning system in the future for detecting as yet unknown viruses with the potential of triggering a new pandemic.
Severe Covid & Phospholipase
Analyzed blood samples from two COVID-19 patient cohorts found that circulation of the enzyme – secreted phospholipase A2 group IIA, or sPLA2-IIA, – may be the most important factor in predicting which patients with severe COVID-19 eventually succumb to the virus.
Early lipidomic studies revealed that severe COVID-19 modifies the circulating lipidome, with decreases in plasma levels of phospholipids and elevated quantities of lysophospholipids (lyso-PLs), unesterified unsaturated fatty acids (UFAs), and acylcarnitines. This lipidomic pattern suggests that severe COVID-19 may be accompanied by cellular or circulating phospholipase(s) that cleave intact phospholipids from cellular and mitochondrial membranes to form lyso-PLs and UFAs.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8483752/
Phospholipases are a ubiquitous group of enzymes that share the property of hydrolyzing a common substrate, phospholipid. Nearly all share another property; they are more active in aggregated substrate above the phospholipids critical micellar concentration (cmc). Phospholipases have low Activity on monomeric substrate but become activated when the substrate concentration exceeds the cmc. Their functions go beyond their role in membrane homeostasis; they also function in such diverse roles from the digestion of nutrients to the formation of bioactive molecules. There are indications that a few phospholipases may carry out a biological function independent of their catalytic activity by binding to a regulatory membrane receptor. Phospholipase-like proteins with toxic properties, which lacked a functional catalytic site, are found in venoms. It is of interest that most, but not all, phospholipases studied in detail thus far are soluble proteins. The soluble nature of many phospholipases suggests that their interaction with cellular membranes is one of the regulatory mechanisms that exist to prevent membrane degradation.
Esterase-Phospholipase balance is important to cmc and balance each other – if esterase dysregulation occurs, perhaps due to excess a7 nAChR activation releasing AChRE into the synaptic cleft, phospholipase will increase to counteract, and may begin degrading the phospho-lipid bilayers making up every cell membrane of every cell type in the body. Like a tailored biological acid the body itself produces.
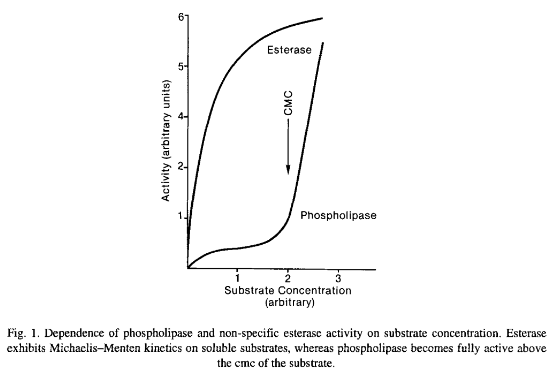
The role of the sPLA2-IIA enzyme has been the subject of study for half of a century and it is “possibly the most examined member of the phospholipase family,” Chilton explained.
Charles McCall, lead researcher from the Wake Forest School of Medicine on the study, refers to the enzyme as a “shredder” for its known prevalence in severe inflammation events, such as bacterial sepsis, as well as hemorrhagic and cardiac shock.
Previous research has shown how the enzyme destroys microbial cell membranes in bacterial infections, as well as its similar genetic ancestry with a key enzyme found in snake venom.
The protein “shares a high sequence homology to the active enzyme in rattlesnake venom and, like venom coursing through the body, it has the capacity to bind to receptors at neuromuscular junctions and potentially disable the function of these muscles,” Chilton said.
Is There Snake Venom in SARS-COV-2?
Anti-idiotype Antibody & Autoimmunity
One way of thinking about the complexity of the immune response is through the lens of anti-idiotype immune responses. The Network Hypothesis, formulated in 1974 by Niels Jerne, described a mechanism by which the antibody responses to an antigen themselves induced downstream antibody responses against the antigen-specific antibody.
Every antibody that is induced and specific for an antigen (termed “Ab1” antibody) has immunogenic regions, particularly in their variable-region antigen-binding domains, that are unique as a result of genetic recombination of immunoglobulin variable, diversity, and joining (VDJ) genes; VDJ recombination results in new and therefore immunogenic amino acid sequences called idiotopes, which are then capable of inducing specific antibodies against Ab1 antibodies as a form of down-regulation.
However, these regulatory immune responses are also capable of doing much more. The paratopes, or antigen-binding domains, of some of the resulting anti-idiotype (or “Ab2”) antibodies that are specific for Ab1 can structurally resemble that of the original antigens themselves (like the spike protein). Thus, the Ab2 antigen-binding region can potentially represent an exact mirror image of the initial targeted antigen in the Ab1 response, and Ab2 antibodies have even been examined for potential use as a surrogate for the antigen in vaccine studies.
However, as a result of this mimicry, Ab2 (Anti-idiotype) antibodies also have the potential to bind the same receptor that the original antigen was targeting. Ab2 antibodies binding to the original receptor on normal cells therefore have the potential to mediate profound effects on the cell that could result in pathologic changes, particularly in the long term — long after the original antigen itself has disappeared.
https://www.nejm.org/doi/full/10.1056/NEJMcibr2113694
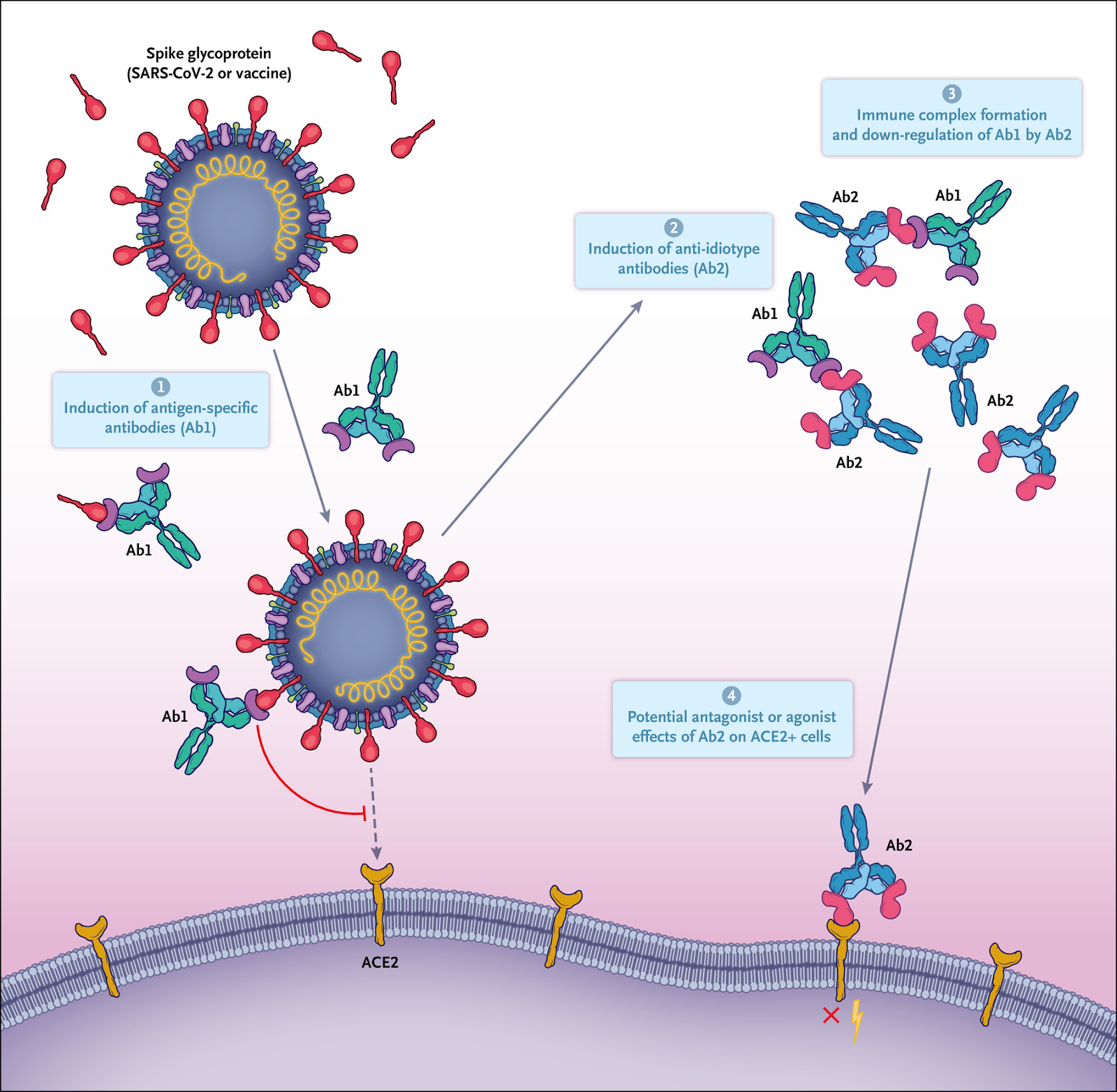
Whereas ACE2 will be the first anti-idiotype to come to mind, it is also important to remember CR3022 and COVA1-16 antibody with the epitope for the recently described a7 nAChR. There is also another sequence of concern, aside from ACE2 and a7nAChR binding amino acid sequences, that we will get into later…
Autoimmune Disease after Infection or Vaccine in Some (Article Review of Anti-idiotype Antibodies)
https://www.nobelprize.org/prizes/medicine/1984/jerne/lecture/
This anti-idiotype mechanism may help explain the Syncytial Lymphocyte Elimination observed in Part 1: “… SARS-CoV-2 spike glycoprotein was identified to be capable of controlling this process by which syncytia, resulted from SARS-CoV-2 infection, could target infiltrated lymphocytes for internalization and CIC (cell-in-cell) mediated death, contributing to lymphopenia in the patients.”
These studies show for the first time that ACE2 antibodies are present after SARS-CoV-2 infection. This finding is consistent with a hypothesis that ACE2 antibodies may be involved in a process that leads to immune activation. While we do not have data about the association of ACE2 antibodies and PASC in this cohort, we hypothesize that antibodies could initiate a cascade of effects that lead to the symptoms of PASC. (A cascade like was shown in Part 1…)
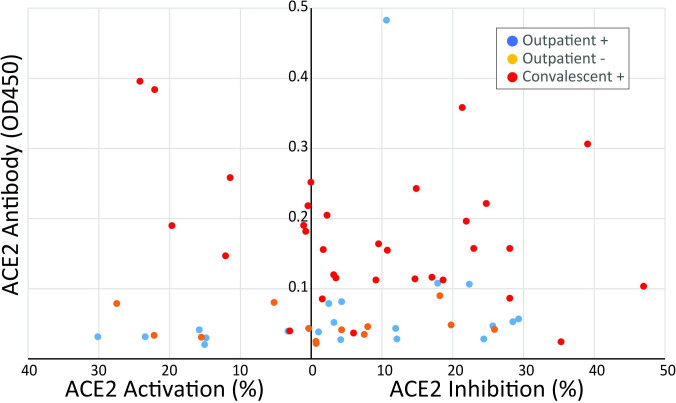
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8415618/
It is important to note that when two antibodies combine, the resulting “immune complex” may circulate until finally settling on a tissue, which will then activate macrophages and cause damage to the tissue or organ they are on.
There are a number of ways to manage this autoimmune response from causing damage. Whereas treatment is NOT a focus of this report, the below video reviews biological mechanisms and potential management approaches.
Autoimmune disease caused by spike protein and potential management approaches
Long Covid & Non-Classical Monocytes
The recent COVID-19 pandemic is a treatment challenge in the acute infection stage but the recognition of chronic COVID-19 symptoms termed post-acute sequelae SARS CoV-2 infection (PASC) may affect up to 30% of all infected individuals. PASC is synonymous with Long Covid.
The levels of both intermediate (CD14+, CD16+) and non-classical monocytes (CD14Lo, CD16+) were significantly elevated in PASC patients up to 15 months post acute infection compared to healthy controls (P=0.002 and P=0.01, respectively). A statistically significant number of non-classical monocytes contained SARS-CoV-2 S1 protein in both severe (P=0.004) and PASC patients (P=0.02) out to 15 months post infection. Non-classical monocytes are capable of causing inflammation throughout the body in response to fractalkine/CX3CL1 and RANTES/CCR5.
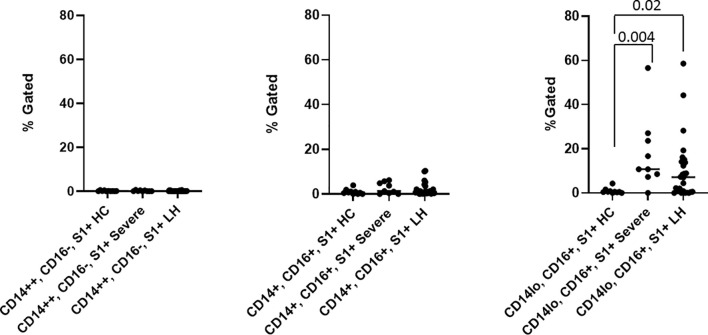
HC, Healthy Control – Severe, Acute Covid – LH, PASC (Long Haul)
Classical monocytes have a circulating lifespan of approximately one day before they either migrate into tissues, die, or turn into intermediate and subsequently non-classical monocytes. Non-classical monocytes expressing high levels of CX3CR1 are involved in complement and Fc gamma-mediated phagocytosis and antiviral responses.
During early stages of the disease, PASC group have reduced classical monocyte and increased intermediate monocyte percentages compared with healthy controls. There is an increase in non-classical monocytes in PASC group 6-15 months post infection, and higher percentages of intermediate and non-classical monocytes at day 0 in severe cases, suggesting augmented classical-intermediate-non-classical monocyte transition in both groups but with different kinetics.
The clinical relevance of monocyte activation in COVID-19 patients and the significance of these cells as viral protein reservoirs in PASC is supported by data reporting the presence of S1 protein within non-classical monocytes. Viral particles and/or viral proteins can enter monocyte subpopulations in distinct ways, and this appears to be regulated differently in individuals that will develop severe disease or PASC. Classical monocytes are primarily phagocytes and express high levels of the ACE-2 receptor.
The hallmark of PASC is the heterogeneity of symptoms arising in a variety of tissues and organs. The CD14lo, CD16+, S1 protein+ monocytes could be preferentially recruited into anatomic sites expressing fractalkine and contribute to vascular and tissue injury during pathological conditions in which this monocyte subset is expanded in non-classical monocytes without S1 protein. Previously, CD16+ monocytes were demonstrated to migrate into the brain of AIDS patients expressing high levels of CX3CL1 (fractalkine) and SDF-1, and mediate blood-brain barrier damage and neuronal injury in HIV-associated dementia via their release of proinflammatory cytokines and neurotoxic factors. Interestingly, a number of papers have been written discussing the increased mobilization of CD14lo, CD16+ monocytes with exercise. These data could help to explain reports of worsening PASC symptoms in individuals resuming pre-COVID exercise regimens.
It is important to note that the S1 protein detected in these patients appears to be retained from prior infection or phagocytosis of infected cells undergoing apoptosis and is NOT the result of persistent viral replication. The S1 subunit itself, while present in non-classical monocytes, is recognized as a CAUSATIVE AGENT responsible for PASC, one of the most chronically severe forms of Long Covid. That is why the US Dept. of HHS has labeled Long Covid a disability under the Americans with Disability Act (ADA).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8784688/
Spike Proteins In Immune Cells – Dr. Bruce Patterson Discusses COVID Long Haul
Dr. Bruce Patterson outlines the pathophysiology of natural and vaccine induced S1 protein caused PASC/Long Covid, biomarker assays to diagnose, and treatment protocols used successfully on 27,000 patients to date.
Dr Bruce Patterson Presentation at Georgetown University on Diagnosis and Treatment of Long COVID
https://pubmed.ncbi.nlm.nih.gov/33186704/
Antibody Dependent Enhancement
In the defense against invading pathogens, Immunoglobulins are formed by B cells. These bind the pathogen via their Fab domains and subsequently activate both complement but also immune cells by immunoglobulin (Fraction constant) Fc-receptors. The largest portion are Immunoglobulin γ (IgG) antibodies that mediate their effector functions through Fc gamma receptors (FcγR)(CD16+) on myeloid (Granulocytes, monocytes, macrophages, and dendritic cells (DCs) represent a subgroup of leukocytes/lymphocytes) and Natural Killer (NK) cells. Antibody dependent cellular cytotoxicity (ADCC) is one of the major Fc-dependent effector functions of IgG, that is particularly important in the clearance of viral infections and is mostly mediated by NK cells. NK cells mediate ADCC through binding of antibody opsonized target cells by membrane expressed FcγRIIIa and induce cytotoxicity by releasing granzymes and perforins stored in intracellular granules.
https://www.frontiersin.org/articles/10.3389/fimmu.2020.00740/full
To block viral attachment to target cells, antibodies that target the viral surface proteins specifically are secreted, which bind and neutralize the viruses. However, in some viruses, the binding of specific antibodies to viral surface proteins can promote viral invasion into certain types of cell instead, and enhance viral infection. This effect is called antibody-dependent enhancement (ADE)
ADE happens in two main cases:
- When viral-specific antibody promotes viral entry into host monocytes/macrophages and granulocytes, and
- When it enhances viral infection in cells via interplay with the Fc receptor (FcR) and/or complement receptor.
Studies so far have presumed that there are five mechanisms that underlie ADE and that various viruses work under different mechanisms and are not necessarily facilitated by a single mechanism.
- The first mechanism of ADE is dependent on FcR. The viral surface protein combines with the antibody to form a virus–antibody complex. The complex strengthens viral adhesion through interaction of the Fc portion on the antibody and its receptor on the surface of particular cells.
- Complement C3 is activated in the classical pathway through the binding of antibody to viral surface protein, following which the interaction between complement C3 and corresponding receptor enhances viral adhesion in the form of virus–antibody–complement complex
- Virus–antibody complexes are combined by C1q, promoting fusion between the viral capsule and cell membrane through the deposition of the combination of C1q and its receptor.
- The fourth mechanism of ADE is the suppression of the expression of antiviral genes via the stimulation and enhancement of certain target cell effects, such as endocytosis, or the suppression of the antiviral genes, such as those for tumor necrosis factor (TNF) and induced nitric oxide synthase (NOS), hence helping the virus with immunological escape.
- The fifth mechanism of ADE is the enhancement of the fusion of viruses and cells via a change in the conformation of viral protein through its binding with antibody (Figure 1E). This was found in HIV infection by Sullivan and co-workers. Monoclonal antibodies recognize the glycoprotein gp120 on the outer membrane of HIV and combine with one of its subunits under a sub-neutralizing concentration. This then induces a conformational change in the residual subunits and promotes membrane fusion of viruses and target cells via the activation of viral glycoprotein. The specific antibody towards gp120 will also regulate the interaction between gp120 and virus ligand CCR5.
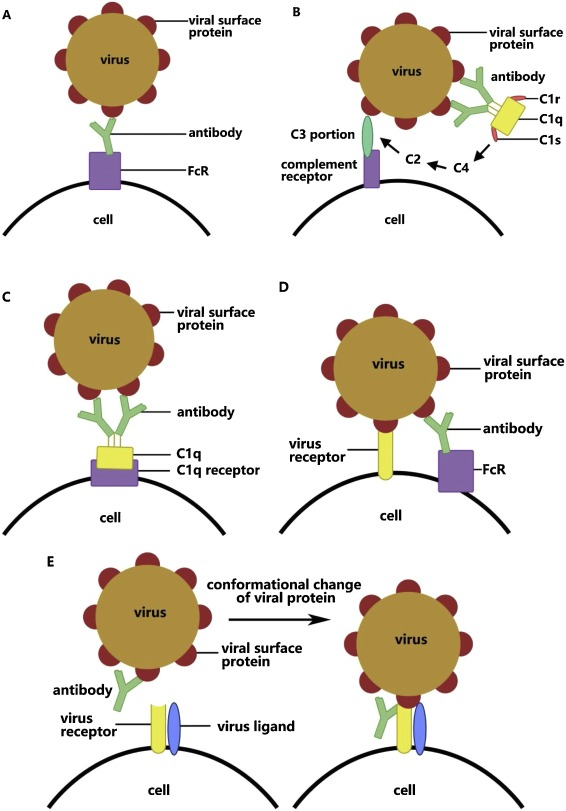
Up until 2019, the mechanism of ADE in SARS-CoV remained unclear. Then Chen et al. developed the first SARS vaccine in 2005, which encodes the complete SARS-CoV viral spike in the modified attenuated poxvirus vector. It was found to induce the production of large amounts of neutralizing antibodies (S-IgG) soon after injection. Although these antibodies can effectively reduce the viral load in the upper respiratory tract, they also enhance lung injury. A positive correlation has been found between the amount of neutralizing antibody in serum and the degree of pathological injury in the lung. Further studies found that the virus enters macrophages with the help of FcR during ADE.
Antibody against SARS-CoV spike alters the function of M2 macrophages (non-classical monocytes) through binding with FcR. Endocytosis of glycoprotein and immunodepression in macrophages are then weakened while the enrichment of cytokines increases. M1 macrophages, which should repair pathological injury in the lung, then convert into cells that promote inflammation. This conversion partially relies on the role of FcγR. *SARS1
https://www.sciencedirect.com/science/article/pii/S1201971220307311#fig0005
CD169+ macrophages have ACE2 and are susceptible to SARS-CoV-2 infection. Both M1- and M2-type macrophages are susceptible to SARS-CoV-2 infection. These observations are likely linked by antibody-dependent enhancement of coronavirus infection of macrophages. The pathophysiology of moderate and severe SARS and COVID-19 diseases fits a proposed model of antibody-dependent infection of macrophages as the key gate step in disease progression from mild to moderate and severe symptoms contributing to dysregulated immune responses including apoptosis for some T cells/T cell lymphopenia, proinflammatory cascade with macrophage accumulation, and cytokine and chemokine accumulations in lungs with a cytokine storm in some patients. Infected phagocytic immune cells may enable the virus to spread to additional organs prior to viral sepsis.
https://www.frontiersin.org/articles/10.3389/fimmu.2021.640093/full
If macrophages become infected, Ivermectin is critical in denying viral load transference into the nucleus for viral replication. No other drug, currently in pharmacology, has exhibited influence on this pathway. It must be approved for those showing elevated humoral response, or antibody dependent enhancement, in order to prevent cytokine storm, Severe COVID, PASC, and Long Covid.
Cytokine Storm with ADE – Antibody-dependent Enhancement of Coronavirus
Non-neutralizing antibodies are what cause ADE. Neutralizing antibodies prevent ADE. What are the implications of continuously inducing vaccine-generated neutralizing antibodies from a two year old strain no longer in natural circulation that wane over time, in an environment of homologous endemic virae, such as coronaviruses?
ADE Pyroptosis
Monocytes and macrophages are sentinel cells that sense invasive infection to form inflammasomes that activate caspase-1 and gasdermin D, leading to inflammatory death (pyroptosis) and the release of potent inflammatory mediators. About 6% of blood monocytes of patients with COVID-19 are infected with SARS-CoV-2. Monocyte infection depends on the uptake of antibody-opsonized virus by Fcγ receptors (ADE Mechanism #1).
SARS-CoV-2 begins to replicate in monocytes, but infection is aborted, and infectious virus is not detected in the supernatants of cultures of infected monocytes. Instead, infected cells undergo pyroptosis mediated by activation of NLRP3 and AIM2 inflammasomes, caspase-1 and gasdermin D.
Moreover, tissue-resident macrophages, but not infected epithelial and endothelial cells, from lung autopsies from patients with COVID-19 have activated inflammasomes. Taken together, these findings suggest that antibody-mediated SARS-CoV-2 uptake by monocytes and macrophages triggers inflammatory cell death that aborts the production of infectious virus but causes systemic inflammation that contributes to COVID-19 pathogenesis.
Increased chronic inflammation is associated with ageing (inflammaging) and the comorbidities linked to severe disease, and severe disease is linked to signs of inflammation. When myeloid cells sense invasive infection, they activate inflammasomes to sound an innate immune alarm. Inflammasome activation is required to process and release interleukin-1 (IL-1)-family cytokines, arguably the most potent inflammatory mediators. However, activation of NF-κB, the TNF receptor superfamily and T helper 17 (TH17) cell cytokines can also cause severe inflammation. When inflammasomes sense infection, they recruit the ASC adaptor and assemble into large complexes that recruit and activate caspase-1, which in turn processes IL-1 pro-cytokines and the pore-forming gasdermin D (GSDMD) to disrupt the cell membrane, leading to cell death and cytokine release. Pyroptotic cell membrane rupture releases cytokines, chemokines and other alarmins that recruit immune cells to infection sites. LDH release is pathognomonic for pyroptosis and other forms of necrotic cell death and elevated LDH is one of the best correlates of severe COVID-19.
https://www.nature.com/articles/s41586-022-04702-4
Summary – ADE in Severe COVID Causing Massive Inflammation
NK Cells & ADCC
While immune responses of the adaptive immune system have been in the focus of research, the role of NK (Natural Killer, CD16+) cells in COVID-19 are extremely important. Antibody opsonization is the process of antibody binding as a marker, and awaiting an Fc-receptor to conduct elimination of the cell. Antibody-dependent cellular cytotoxicity (ADCC) is the process after recognition, by which phagocytes envelope, or NK cells attack with perforins and granzymes. Perforins perforate the cell membrane, granzymes, stored in granules, are then inserted to “digest” the cell.
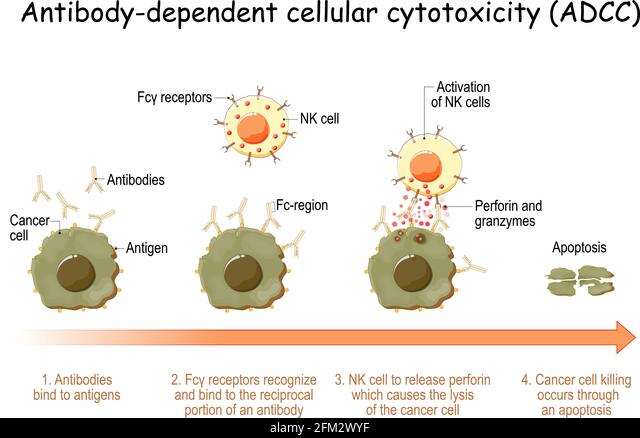
Serum samples from SARS-CoV-2 resolvers induced significant CD107a-expression by NK cells in response to S1 and NC, while serum samples from SARS-CoV-2-negative individuals did not. Serum samples from individuals that received the BNT162b2 vaccine induced strong CD107a expression by NK cells that increased with the second vaccination and was significantly higher than observed in infected individuals.
S1-specific CD107a responses by NK cells were significantly correlated to NK cell-mediated killing of S1-expressing cells. Interestingly, screening of serum samples collected prior to the COVID-19 pandemic identified two individuals with cross-reactive antibodies against SARS-CoV-2 S1, which also induced degranulation of NK cells.
Taken together, these data demonstrate that antibodies induced by SARS-CoV-2 infection and anti-SARS-CoV-2 vaccines can trigger significant NK cell-mediated ADCC activity, and identify some cross-reactive ADCC-activity against SARS-CoV-2 by endemic coronavirus-specific antibodies.
https://www.nature.com/articles/s41598-018-36972-2
Natural Killer Cells and Antibody Dependent Cellular Cytotoxicity (ADCC)
Chronic Inflammation & Cytokines
Inflammation is the principal process used by the innate arm to combat & clear out microbes or damaged cells due to; infection, injury, cancer, environmental irritants, toxic substances, sui pathogenesis, etc.
When there is a continual process of inflammation then slowly the area damaged may get scarred with calcium or amyloid deposits, leading to permanent damage. All tissues have sentinel (sensor) cells, such as Dendritic cells, macrophages, and mast cells. Sensor cells within the tissue have multiple functions:
- Diagnose origin of tissue damage
- Engage with foreign substance
- Signal additional responses
Liver = Kupfffer’s cells, Spleen = Splenic macrophages, Nervous = MicroGlia, Lungs = Alveolar Macrophages (Dust cells), Peritoneum = Peritoneal Macrophages, Epithelial (Skin) Cells = Langerhans cells; are system specific sentinel cells of the innate arm of immunity.
Toll-like Receptors on sentinel cells, have two forms:
- Damage Associated Molecular Pattern (DAMP)
- Pathogen Associated Molecular Pattern (PAMPs)
Cellular material will initiate a response from DAMP. Foreign substances, such as microbes, bacteria, and pathogens, trigger PAMPs. The activation of toll-like receptors (DAMP, PAMPs) in sentinel cells will cause the production of cytokines. If doctors understand the pathologies associated with cytokines present in the system, they will be better able to diagnose the nature of disease, and formulate treatment solutions.
TNF, (Interleukin) IL-1, IL-6, IL-10, IL-12, IL-15, IL-18, IL-23, IL-27, IFNα,β are all types of Cytokines. There are more.
Chemokines are specialized Cytokines principally resulting in cellular recruitment to a site:
- CCL2, 3, 4, 5, 11, 17, 19, 21, 22, 25, 27
- CXCL 1, 8, 9, 10, 12, 13
- XCL1 (Lymphotactin)
- CX3CL1 (Fractalkines)
TNFα, IL-1, & IL-6 being the most critical. Most Cytokines operate in a localized paracrine action. However, it is possible for them to be released into the bloodstream and operate as an endocrine, possibly causing a pathogenic cascade, anywhere connected to the system.
For example: Interleukin has been shown to produce acute phase reactant proteins in the Liver. Cytokines in the Brain can affect temperature regulation in the hypothalamus. A humoral response may be triggered in the bone marrow. These are all distinct examples of what may happen when a disease enters a chronic phase, and release of cytokines cascades into other pathologies downstream of tissue undergoing infection, apoptosis, pyroptosis, etc.
Cytokines have Three primary functions:
- Regulate Local Innate/Inflammatory response
- Inhibit Viral Replication
- Activate Adaptive Arm of Immune System
The sequence of events of inflammation:
- TLR/PRR/DAMP/PAMP sensory receptors detect damage or pathogens, and begin releasing cytokines.
- Permeability of nearby capillaries is increased and dilated to open channels to areas of infection/damage.
- Blood flow increases in the area and Antibodies, Collectins, Complement proteins, pentraxins, etc begin flowing into the site.
- TNF & IL-1 act on epithelial cells to begin producing ligands. Ligands (Integrins, E-Selectins, ICAM-1, VCAM-1, etc) are specialized signal receptors for White Blood Cell recruitment from blood vessels to affected tissue.
- White Blood Cells release cell adhesion molecules to bind with ligands formed on the blood vessel.
- Transmigration occurs when the ligand fuses with the cell adhesion molecule and the White Blood Cell creates a path from the blood vessel to enter the affected tissue.
- White Blood Cells then become inflammatory-infiltrate and begin working in the affected area.
Chronic Inflammation (Lecture 1)
Phagocytosis & Chronic Inflammation
Phagocytosis is the process of a cell internalizing (eating, surrounding, etc.) a pathogenic substance for elimination. While in a normal resting state phagocytes (macrophages and neutrophils) are NOT producing cytokines or actively engaged in elimination. Once activated after DAMP/PAMP receptor binding, cytokines will be released and the phagocyte will envelop the microbe.
In order for phagocytosis to occur, the pathogen/microbe will need to be opsonized with antibodies. Fc Receptors on the phagocyte bind with Fractional constant proteins of the antibody. Complement factors, such as C3b, produced by the liver as an acute phase reactant protein, may also bind to the microbe in order to opsonize it just like an antibody.
When a pathogen is internalized by a phagocytosis, it will be held in a vesicle called a Phagolysosome. Lysosomes, enzyme packed vesicles within the phagocyte, will then fuse with the Pathogenic phagolysosome and release its enzymatic contents. Reactive Oxygen Species (ROS), critical in chronic inflammation, are made to a greater extent in neutrophils, Nitric Oxide is made to a greater extent in macrophages. Inducing Nitric Oxide Synthase (iNOS) converts arginine to citrulline in order to produce NO. Oxidase producing ROS are also activated by IFN-γ and TLRs(DAMP/PAMP). If ROS and NO are unable to degrade the pathogen inside the phagolysosome, the macrophage will become “frustrated” and may self-select to Pyroptosis, leading to the release of inflammatory cytokines into the body.
Chronic Inflammation – Pathogen Killing – Phagocytosis (Lecture 2)
Immune Regulation & Chronic Inflammation
Immune system regulation between the innate and adaptive (acquired) arms of immunity are meant to Amplify each other until a pathogen or disease is eliminated, as well as to return back to homeostasis when the threat has been dealt with. Regulation helps prevent excessive activation and protect from the immune system attacking on the body. The duration, location, and intensity of the immune response need to be carefully coordinated. If immune cells begin unnecessarily attacking our own body then it may lead to an autoimmune induced state of chronic inflammation.
Antigen tolerance development, immune exhaustion, genetic factors, memory cells, and molecular mimicry all play a role in the speed and nature of response, as well as duration of activity.
The adaptive arm of immunity begins when the innate arm (macrophages & dendritic cells) present a protein from the pathogen (antigen) to “Naive” CD4+ T-helper cells. If IL-4 is present and IL-12 is absent, then the “Naive” CD4+ T-helper cell will convert down the Th2 pathway, or humoral response, and later convert into a B-Cell. If IL-12 is present, then the T-helper cell will go down the Th1 pathway, and later convert to a cytotoxic CD8+ Killer T-Cell.
Th17 and T-Reg cells are T-helper cells mainly geared toward regulating the response of both the innate and adaptive arms of immunity. IL-10 is an immuno-modulator of inhibition or deactivation of the innate arm.
Suppressors of Cytokine Signaling (SoCS) receptors are formed after innate arm activation of TLR (DAMP/PAMP), on the cell itself. SoCS receptors are presented as a feedback mechanism for the cell to deactivate the cell with an inhibitory cytokine, such as IL-10. SoCS receptors, if activated, will inhibit the Jak-stat signal pathway in the cell from functioning if the cell’s cytokine receptors are activated, thus suppressing the production and influence of cytokines within the cell. Every single cell has its own regulatory mechanism.
IFN-γ, when released by Th1 cells, will upregulate the innate arm and increase inflammation by raising the activity of major histocompatibility complexes (MHC). MHC-2 is required for the innate arm to present an antigen to the adaptive immune cells. The more antigens to present, the higher the inflammation.
If a B-cell becomes active in generating antibodies after an antigen binds to one of its antigen receptors, it may become down-regulated if an Fc on an antibody-antigen complex binds to an FcγRIIβ on the B-cell at the same time as the antigen it is attached to. In this case, ITim would be activated in the B-cell, thus inhibiting any further antigen binding. If there are any mutations on either the Fc or the FcR preventing binding, then there will be no inhibitory signal to stop production of antibodies within the B-Cell.
Chronic Inflammation – Immune System Regulation (Lecture 3)
https://www.cell.com/cell/fulltext/S0092-8674(20)31542-7
Cytokines & Neurodegeneration
The dysregulation of cytokines and chemokines is a central feature in the development of neuroinflammation, neurodegeneration, and demyelination both in the central and peripheral nervous systems and in conditions of neuropathic pain. Pathological states within the nervous system can lead to activation of microglia.
The latter may mediate neuronal and glial cell injury and death through production of proinflammatory factors such as cytokines and chemokines. These then help to mobilize the adaptive immune response.
Although inflammation may induce beneficial effects such as pathogen clearance and phagocytosis of apoptotic cells, uncontrolled inflammation can result in detrimental outcomes via the production of neurotoxic factors that exacerbate neurodegenerative pathology. In states of prolonged inflammation, continual activation and recruitment of effector cells can establish a feedback loop that perpetuates inflammation and ultimately results in neuronal injury.
A critical balance between repair and proinflammatory factors determines the outcome of a neurodegenerative process.
In the field of neuroimmunology, the classical view that regarded the central nervous system (CNS) as an immune-privileged site by virtue of its shield, the blood brain barrier (BBB), has evolved to a view of significant CNS-immune system interactions.
Cytokines and chemokines are involved in the regulation of CNS-immune system interactions besides being important for the coordination of immune responses throughout the body. They are produced primarily not only by white blood cells or leukocytes but also by a variety of other cells as a response to various stimuli under both pathological and physiological conditions. In the nervous system, cytokines and chemokines function as neuromodulators and regulate neurodevelopment, neuroinflammation, and synaptic transmission.
This review will focus on how cytokines and chemokines affect neuroinflammation and disease pathogenesis in bacterial meningitis and brain abscesses, Lyme neuroborreliosis, human immunodeficiency virus encephalitis, and neuropathic pain.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3753746/
Sequence of concern: FCS/PRRA/MSH3
Among numerous point mutation differences between the SARS-CoV-2 and the bat RaTG13 coronavirus, only the 12-nucleotide furin cleavage site (FCS) exceeds 3 nucleotides. A BLAST search revealed that a 19 nucleotide portion of the SARS.Cov2 genome encompassing the furin cleavage site is a 100% complementary match to a codon-optimized proprietary (patented) sequence that is the reverse complement of the human mutS homolog (MSH3).
While numerous point mutation differences exist between SARS-CoV-2 and RaTG13, only one insertion and dissimilarity exceeding 3 nucleotides (nt): a 12-nucleotide insertion coding for four amino acids (aa 681-684, PRRA) in the SARS-CoV-2 S protein has been discovered. This polybasic FCS differentiates SARS-CoV-2 from other b-lineage betacoronaviruses or any other sarbecovirus. An FCS addition enhanced the infectivity of SARS Co-V-2 in 2019. The absence of this FCS results in attenuated SARS-CoV-2 variants useful for animal vaccination, accentuating its relevance to human infection. This FCS is vital for human and ferret transmission, expands viral tropism to human cells, and is requisite for severe disease in two animal models of SARS-CoV-2.
A BLAST search for the 12-nucleotide insertion led to a 100% reverse match in a proprietary sequence (SEQ ID11652, nt 2751-2733) found in the US patent 9,587,003 filed on Feb. 4, 2016. It is important to note that gene do NOT have to be an exact match in order to produce an identical protein. Codons are sequences of 3 genes producing a protein. I have used the term “homologous” in the past, and that is to indicate relation to two proteins, even though the codons (genes) producing them may be different.

The proprietary sequence SEQ ID11652, read in the forward direction, encodes a 100% amino acid match to the human mut S homolog 3 (MSH3). MSH3 is a DNA mismatch repair protein (part of the MutS beta complex). SEQ ID11652 is transcribed to a MSH3 mRNA that appears to be codon optimized for humans. We did not find the 19-nucleotide sequence CTCCTCGGCGGGCACGTAG in any eukaryotic or viral genomes except SARS-CoV-2 with 100% coverage and identity in the BLAST database.
Overexpression of MSH3 is known to interfere with mismatch repair (MSH2 sequestration from the MutS alpha complex comprising MSH2 and MSH6 results in MSH6 degradation and MutS alpha depletion), which holds virologic importance. Induction of DNA mismatch repair deficiency results in permissiveness of influenza A virus (IAV) infection of human respiratory cells and increased pathogenicity. Mismatch repair deficiency may extend shedding of SARS-CoV-2.
https://www.frontiersin.org/articles/10.3389/fviro.2022.834808/full#B15
MSH3, MSH2, and MSH6 are responsible for DNA repair. If there is an imbalance of any of these genes then there may be disruption, repair inhibition, or mutation of the host genome. At a point of genetic damage, genetic repair will occur or the cell will lose function. The primary genetic respondents to genetic damage are MSH2 & MSH6. MSH3 is known to bind to MSH2. If MSH2 is not available to bind with MSH6 for genetic repair, then genetic dysregulation may occur.
An investigation must be conducted. MSH3 leads to more severe disease, longer recovery times, and is carcinogenic. It is a genetic pathogen.
Spike Genes Have Patented DNA Sequences. This is Dangerous.
The protein encoded by this MSH3 forms a heterodimer with MSH2 to form MutS beta, part of the post-replicative DNA mismatch repair system. MutS beta initiates mismatch repair by binding to a mismatch and then forming a complex with MutL alpha heterodimer. This gene contains a polymorphic 9 bp tandem repeat sequence in the first exon. The repeat is present 6 times in the reference genome sequence and 3-7 repeats have been reported. Defects in this gene are a cause of susceptibility to endometrial cancer.
https://www.ncbi.nlm.nih.gov/gene/4437
At this moment the idea of inducing MSH2 production may seem advisable to counteract an abundance of MSH3. However, the presence of MSH2 implies damage and usually induces the creation of cytokines. MSH3, in this case, is a patented human optimized genetic codon sequence, meant for oncology (cancer) related proteins and peptides, that occured naturally in a wet market…
PRRARS
Scientists scanned the human databases (Taxon 9606) and found that the exact sequence PRRARS was present only in two human proteins:
- AAB17869.1, Hermansky-Pudlak syndrome protein (HPS1) – Hermansky-Pudlak syndrome (HPS) is a rare, hereditary disorder that consists of two characteristics: decreased pigmentation (albinism) with visual impairment, and blood platelet dysfunction with prolonged bleeding. Some patients have lung fibrosis, colitis, or an abnormal storage of a fatty-like substance (ceroid lipofuscin) in various tissues of the body.
https://rarediseases.org/rare-diseases/hermansky-pudlak-syndrome/ , and
- AAF79955.1, RhoGEF (RhoGEF domain is a structural domain of guanine nucleotide exchange factors for Rho/Rac/Cdc42-like GTPases. It is also called “Dbl-homologous” (DH) domain.)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7833556/#appsec1
Genetically Toxic Pathogen
I did not think I would find a patented protein when this began. Much less a carcinogenic human codon optimized MSH3 gene that could cause genetic repair dysregulation. I believe PRRA on the SARS-CoV-2 Spike protein furin cleavage site (FCS) fits the definition of a genetic toxin.
I can only imagine the wisdom in artificially creating the spike protein intracellularly with human optimized mRNA known to dysregulate genetic repair, with a technology capable of infiltrating monocytes for reproduction. ADE was already found to be an issue with this spike protein, but might as well give them a head start…
Those saying the spike protein has been modified with primers and prolines to prevent any of this from happening are lying. 2 prolines have been added to keep the S2 subunit in a prefusion/closed state ONLY. There have been NO MODIFICATIONS when it comes to the S1 subunit, which is the therapeutic target. If S1 is the therapeutic target then it MUST be cleaved from S2 thus exposing the most pathogenic part of SARS-CoV-2 within the cell. Imagine also anti-idiotype antibodies expressing PRRA/MSH3…
Part 3 will be all about vaccines. Vaccines attempt to stimulate your immune system, and you now have full context of the immune mechanisms involved with generating the antibodies necessary to “combat COVID19”. You also have full knowledge of the toxic structure and effects. What we don’t know are every possible mutagenic consequence of its application.
In this sense “Long COVID”, outside of ADE S1 infected monocytes and chronic inflammation, may even be hereditary.
https://www.nature.com/articles/s41541-021-00369-6
S1 is a toxin

Leave a comment